

- Help
- General Tips
- Job Options
- Plot Options
- Advanced Options
- Circular Dichroism Calculation
- Linear Dichroism Calculation
- Job Submission
- Protein Sizes and Compiler Limits
- File Types
- Results
Help
DichroCalc aims to make Dichroism calculations available to a wider range of scientists than a small number of computational chemists. Without being exposed to Linux and cryptic command-line switches, it is possible to choose from a number of different parameters and calculate CD and LD spectra just by ticking boxes. To date, DichroCalc is the only interface available offering circular and linear dichroism calculations.
The theory behind the calculations was published in PCCP and it is highly recommended to read into the background of the methods. Using theoretical chemistry as a black box method bears its dangers and knowing how the calculations are carried out helps for example to analyze the log files to verify whether the groups of the protein have been recognized correctly.
General Tips
It was tried to keep the usage of DichroCalc as easy as possible. However, to benefit most from its use it is strongly recommended to read through this help section. This is especially important or the options that are used for a calculations. Next to each option on the interface the is a Help small Help link (see to the right) that leads directly to the respective section in this help file.
A common mistake that should be avoided in general in computational chemistry can be avoided with one simple rule:
Start with a plain calculation.
"Plain" means a standard calculation without bells and whistles. This is crucial for several reasons:
- If the calculation with the basic parameters takes long, you are prepared that additional options may take much longer.
- If a certain feature is found when, for example, charge-transfer parameters are used, one has to run the plain calculation anyway in order to verify that the origin of the feature are indeed charge-transfer transitions.
- Especially if the protein is quite large (mind the section on protein sizes), considering chromophores in addition to the peptide groups may exceed the number of groups that can be considered due to compiler limits.
Do not simply tick all available chromophores, especially if they are not contained in the protein anyway. This will cause the calculation to fail if the maximum number of types is exceeded.
Job Options
Uploading Files
It is possible to upload a number of PDB files at once by combining them in an archive. Currently the following archive types can be handled:
- .pdb
- .tar, .tar.gz, .gz, .tgz
- .zip
- .Z
Uploading an archive enables the user to run calculations on a batch of PDB files at once. For uploading experimental spectra please see further down.
PDB Input Files
The input files for DichroCalc are .pdb files according to the standards of the RCSB Protein Data Bank. Important for the files to be interpreted correctly is the ATOM section (the header and footer information is discarded as are the HETATM lines). Particularly the positions of the atom types, residues and coordinates need to be correct. In each residue, the atoms required for using the respective parameters are searched and only if all are found the parameters are used for this group. The following atom types are essential for the correct assignment of the respective chromophore atoms. Note that there must not be ambiguities, that is if more than one of those atom labels is found within a residue, it is ignored.
- Peptide-Backbone:
C, N, O
- Charge-Transfer transitions:
These chromophores are made up from two adjacent backbone peptide bonds, i.e. twice the C, N, O atoms. Gaps within a PDB chain due to missing residues are accounted for. - Side chain transitions:
- Phenylalanine:
CG CD1 CE1 CZ CE2 CD2 - Tyrosine:
CZ CE1 CE2 CD1 CD2 CG OH - Tryptophan:
CG CD2 CE2 CE3 CD1 NE1 CZ2 CZ3 CH2
- Phenylalanine:
- Nucleic bases:
- Adenine (A):
N1 C2 N3 C4 C5 C6 N6 N7 C8 N9
- Guanine (G):
N1 C2 N2 N3 C4 C5 C6 O6 N7 C8 N9
- Thymine (T):
N1 C2 O2 N3 C4 O4 C5 C6
- Cytosine (C):
N1 C2 O2 N3 C4 N4 C5 C6
- Uracil (U):
N1 C2 O2 N3 C4 O4 C5 C6
- Adenine (A):
- Naphthalenediimide (NAP):
O1 N1 O4 C3 C10 C4 C9 O2 N2 O3
- Oligoureas (URE):
N' O N
- Peptide bonds between non-adjacent residues:
Currently cyclic peptide bonds between the side chains of lysine and aspartic acid are supported. If this option is selected, the distances between the respective atoms are calculated and regarded as a peptide bond if they are within bonding distance. The atom labels are:
CG OD1 NZ
PDB files can contain multiple conformations of the same structure (using MODELs). DichroCalc can recognize the different structures and always extracts the first model from the PDB to calculate it. If the spectrum of a different model than the first one is required, the PDB has to be split up into individual files. The PDB header information is actually not required and it is sufficient to just copy each MODEL into a new file. Using the Perl library ParsePDB.pm, this can be easily automated:
use ParsePDB; $PDB = ParsePDB->new (FileName => "foo.pdb"); $PDB->WriteModels;
The PDB file is checked for alternative atom locations (of which the first one is kept) and inserted residues. If the latter are found, they are kept unless they are superpositions of each other (like an alanine superimposed by an isoleucine, which would then be an alternative residue). In case of superpositions, the residue with an inserted residue tag is removed.
The correct atom labels for each residue can be found in the Chemical Component Dictionary by selecting the respective residue and clicking on "Chemical diagram with hydrogen atom labels" at the bottom of the page.
Uploading Experimental Spectra
DichroCalc returns an archive containing the requested spectra as xy values for the use in a spreadsheet application and additionally plotted as a postscript file. It is also possible to obtain a comparison plot of the experimental spectrum with the calculation.
The experimental spectrum needs to have the same base name as the corresponding PDB file with the
correct extension for the respective spectrum type (e.g. 4mbn.pdb and 4mbn.exp.cd). The
extensions are given below.
Retrieving Files from the RCSB PDB
Instead of uploading a file from the local machine, the PDB files can also be retrieved via their PDB code from the RCSB Protein Data Bank. Simply enter the PDB codes separated by spaces or commas.
PDB codes can be given on their own or in addition to an uploaded PDB file or an archive.
Job Name
The name of the job is usually made up of the uploaded file name. Alternatively it is possible to define a custom job name, which will be turn up in the name of the output directory sent to the user and the main log filename of the job.
Charge Transfer Transitions
Charge-transfer transitions have an influence on the spectrum in the deep UV below 190 nm. Two peptide groups form one charge-transfer chromophore (see protein sizes) and add four states to the calculation. Therefore, adding charge-transfer chromophores considerably lengthen the calculation time.
Aromatic Side Chain Chromophores
By default only the backbone chromophores are considered in the calculation. The chromophoric groups of the side chains dominate the CD spectrum in the near-UV (250 nm - 350 nm). If the checkbox is ticked, the chromophoric groups of phenylalanine, tyrosine and tryptophan are also taken into account. Remember to extend the wavelength range to cover the near-UV. For large proteins please mind the section on protein sizes and compiler limits.

If the checkbox ( with the associated vibrational structure ) is ticked, vertical transitions to the various vibrational levels of the electronic excited states will be considered (modelled via the Franck-Condon factors). Transitions appearing in the near-UV (250 nm - 350 nm) are the 1Lb transition of PHE, TYR and TRP and 1La of TRP. Considering the vibrational structure of excitations will noticeably increase the size of the exciton Hamiltonian matrix.
Non-aromatic Side Chain Chromophores
Via these checkboxes the peptide bonds contained in the side chains of asparagine, aspartic acid, glutamine, and glutamic acid can be taken into account.
Some side chain groups can form peptide bonds with each other which leads to cyclic peptides, i.e. bonds between non-adjacent residues. Currently the side chains of lysine and aspartic acid are supported (atom types CG OD1 NZ), more combinations can be made available on request.
Nucleic Bases
The parameters of the nucleic bases adenine, guanine, cytosine, thymine, and uracil allow calculation of DNA and RNA CD and LD spectra. Each base can be selected or omitted individually.
The PDB residue labels A, G, C, T, and U are required (some PDB files were found to use different labels and those need to be corrected).
To create DNA/RNA PDB structures from a given sequence in A or B conformation, James Stroud's Make-NA server is much recommended.
Naphthalenediimide
For the consideration of the naphthalenediimide (NDI) chromophore it is particularly crucial to verify the correct atom labels:
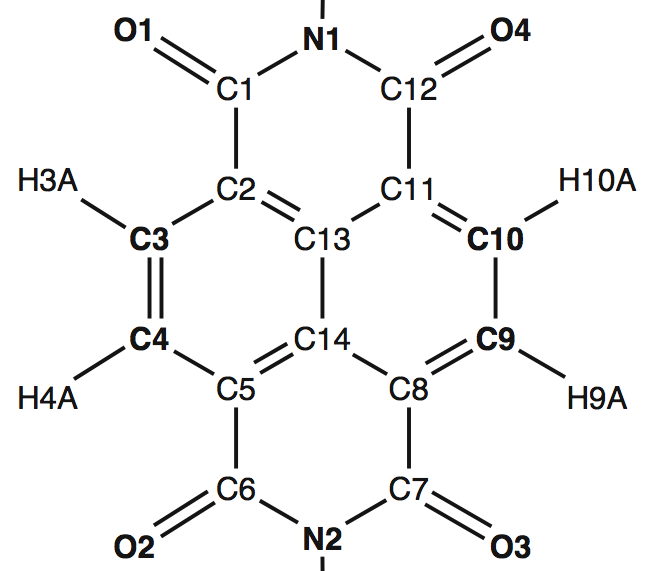
The atoms shown in boldface are used to determine the orientation of the chromophore. It was found that the standard bandwidth of 12.5 nm is too broad to resolve the spectral features and a bandwidth of 6 nm is recommended.
Oligoureas
By default, the input files are assumed to be oligo- or polypeptides and the respective chromphores are assigned to the backbone. If urea parameters are to be assigned to the backbone chromophores, this has to be selected under Advanced Options ⇒ Parameter set for the backbone chromophores. The default number of backbone transitions for urea chromophores is 3 and the dropdown box will change to this number upon selection of the urea parameters. After the selection, the number of backbone transitions can be changed to 1–4 (choose 0 transitions to ignore the backbone).
For a correct assignment of the parameters, the urea residues must have a URE label (also see PDB Input Files).
Mind that it is at present not possible to consider peptide and urea chromophores at the same time.
Calculated Range Plot Options
Plot Spectra as Postscript files
All calculated spectra are automatically plotted as .ps files (including the experimental
spectrum, if provided). If you submit a large amount of spectra (like a run of MD simulations
for example), it is recommended to turn the automatic plotting off.
Range and Interval of the Wavelengths
By default the wavelength range of 150 to 350 nm is calculated every nm. If another range is required, e.g. up to 450 nm, or a smaller interval is needed, it can be selected in this section.
Unless the tick box to plot all spectra is deselected, all calculated results are plotted as PostScript files. The effect of the selected chromophores may differ by orders of magnitude and wavelength region (for example protein backbone and aromatic side chains). In such cases it is necessary to magnify the low intensity region and this can be achieved by defining a second region to plot. In this case, two plots will be generated for each spectrum.
Advanced Options
Average Spectra
This option triggers the calculation of averaged spectra for each calculated spectrum type (CD, LD, dichroic ratio, reduced LD).
Mind that individual PDB files are required and only the first MODEL of each PDB files is calculated.
Group Interaction Analysis
Please note: This feature is computationally very expensive and may take a long time to process, producing several hundreds of megabytes. Please use it considerably, that is only if the (already available spectra) indicate that further analysis is necessary.
The group interaction analysis performs an analysis of the Hamiltonian matrix and lists the interactions between all
transitions on all groups (i.e. chromophores) in the .stat file, a tab-separated
text file, which can be imported into a spreadsheet application.
The interactions are given in wavenumbers, the distances between the chromophores in Angstrom:
| Res1 | Group1 | Trans1 | Assignments | Res2 | Group2 | Trans2 | Assignments | Distance | Interaction |
| 1 | 1 | nPi* | 3 4 12 | 1 | 1 | PiPi* | 3 4 12 | 0.00 | -11.2965 |
| 1 | 1 | nPi* | 3 4 12 | 2 | 2 | nPi* | 14 15 20 | 3.41 | 95.4737 |
| 1 | 1 | nPi* | 3 4 12 | 2 | 2 | PiPi* | 14 15 20 | 3.41 | -17.7169 |
| 1 | 1 | nPi* | 3 4 12 | 3 | 3 | nPi* | 22 23 26 | 6.68 | 0.4481 |
| 1 | 1 | nPi* | 3 4 12 | 3 | 3 | PiPi* | 22 23 26 | 6.68 | -3.5746 |
The residue numbers, Res1 and Res2, refer to the renumbered PDB file, which is included in the .log file of the calculation. The chromophores, Group1 and Group2 are numbered sequentially as they are assigned during the calculation, first the peptide groups, then the charge-transfer chromophores and finally the different side chain groups. The group assignments are the atom numbers defining the chromophore. Group1, for example, is usually the peptide bond between the first and second amino acid and is defined by atoms 3, 4 and 12 in the above example, which are the atoms C and O of residue 1 and atom N of residue 2. The distances are the distances between the first two assigned atoms. It is recommended to crosscheck the output given in the .stat and .log file, especially the atom assingments.
Parameter Sets for the Backbone Chromophores
It is possible to choose from four different parameter sets for the backbone chromophores. The first two are peptide paramenters, of which one is derived from ab initio calculations and one from semiempirical methods.
The third parameter set is for dimethylurea to calculate spectra for oligoureas. It was derived from ab initio calculations.
The fourth choice is the peptide parameters considering the vibrational structures of the π→π* transitions. It was derived from ab initio calculations.
Number of Backbone Transitions
By default, for the peptide parameters two backbone transitions are considered, which are the n→π and π→π * transitions. By increasing this number, it is possible to include higher energy transitions, the πb→π* (bonding π orbital to π*) and n'→π* (second lone pair on oxygen to π*). The latter two transitions occur around 140 nm.
More than two transitions are only available for the ab initio parameter set for CD calculations.
If the peptide with vibrational parameter is selected, you can choose up to 6 transitions here to take into account 1 n→π* transition and 5 π→π* transitions.
By selecting 0 transitions the backbone can be completely ignored, for example, to analyze only the side chain contributions or the nucleic base moiety of a protein containing a DNA strand.
Curve Types
This selects the bandshape of the curves which are fitted to the line spectrum to mimic the broadening of the bands in solution.
In an experimental spectrum, the transitions are broadened due to the uncertainty principle, unresolved vibronic components and the interaction of the chromophore with its environment including other chromophores and the solvent. Thus, an overlay of approximately Gaussian shaped bands is observed. Since the result of the calculation is a line spectrum, a convolution with a lineshape function is required.
The curve types can be chosen from approximate Lorentz shape, Lorentz and Gaussian shape; the latter gives better results than Lorentzian curves.
Bandwidth at Half Maximum
This defines the width of the bands which are superimposed over the line spectra. The default is 12.5 nm which was shown to be the best value. Do not change this unless you know exactly what you are doing.
Circular Dichroism Calculation
Units
This dropdown field defines the units of the returned spectra (in the xy data files as well as in the postscript files). It is possible to choose between ellipticities (deg cm2 dmol-1) and absorbance units (mol-1 dm3 cm-1). The original output units of the calculation are ellipticities. If absorbance units are selected, the intensities are divided by 3300. Choosing the output units is particularly important if you provide experimental spectra. If these are given in absorbance units but ellipticities are selected, the comparison plots are useless, although it does not harm the calculation itself.
Full CD Analysis
Please note: This feature is computationally very expensive and may take a long time to process. Please use it considerably, that is only if the spectra indicate that further analysis is necessary.
The full CD analysis provides a possibility to comfortably run all possible combinations of the chosen parameters on the input files. If, for example, side chains and charge-transfer is selected the routine will return:
- backbone-only;
- including charge-transfer transitions;
- including all side chain chromophore (with and without CT)
- including every single side chain chromophore on its own (with and without CT);
Only the parameters, which are actually needed should be selected since unnecessary calculations waste time and will bloat the comparison plots, thereby complicating the analysis. Adding an additional backbone transition in the deep-UV, for example, would double the amount of calculations since everything is done for two transitions (the default) and three transitions. Likewise for a fourth transition.
Along with all spectra files, a report is returned as PDF, showing several comparison plots of the spectra. If an experimental spectrum was included, it is automatically included in the plots.
Linear Dichroism Calculation
The dependence of protein orientation presents an additional challenge for the linear dichroism calculation with respect to CD.
Aligning the Protein
Before performing an LD calculation it is important to align the molecule to the desired position. DichroCalc uses the following definitions to calculate the dichroism:
- x-Axis: perpendicular
- z-Axis: parallel
To align the PDB, the easiest tool to use is the freeware Swiss-PdbViewer which can be downloaded at www.expasy.org/spdbv and is available for Windows, Linux and MacOS.
Open Swiss-PdbView and load the PDB you want to align. It will be displayed in the same orientation as it is defined in the PDB file (actually you can also load mmcif and mol files). Click in the tool bar on Display => Show Axis to display a small coordinate system in the top left corner. It will be oriented in "arbitrary" position defined by the coordinates in the PDB file:
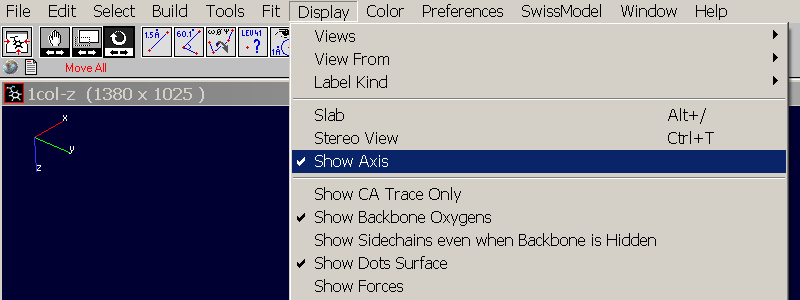
Select the "Rotation Tool"  and
rotate the molecule until the z-axis is oriented straight up and the x and y axes are
shown exactly perpendicular to it:
and
rotate the molecule until the z-axis is oriented straight up and the x and y axes are
shown exactly perpendicular to it:

These rotations were done in the "Move All"-mode, which is selected by default and translates the molecule as well as the coordinate system. To rotate only the molecule but keep the coordinate system as it was just oriented, one has to switch the "Move Selection"-mode by clicking on the respective red text-button on the lower left corner of the toolbar:

Now the coordinate system is fixed and only the selected atoms are rotated. To be sure that the whole protein is selected click Select => All or press <Ctrl>-<A>. Rotate the molecule to its desired oritentation (usually this will be the long axis aligned to the z-axis) and save the new coordinates with File => Save => Layer.
Depending on the overall shape of the protein, it is sometimes easier to have the z-axis pointing towards the user, that is x and y oriented parallel to the screen. Aligning the protein to the z-axis means then minimizing the visible surface of it.
Automating the Search for the Correct Orientation
DichroCalc offers an automated calculation of LD spectra for a series of different orientations of the protein. This "orientation search" can be requested by ticking the repective tick box in the form. For this run, two axes need to be defined:
- The tilt axis should be perpendicular to the molecule axis. Around this axis the molecule is rotated about a defined tilt interval until a maximum tilt angle is reached. DichroCalc assumes the protein to be oriented with its long axis to the z-axis (parallel direction), by default the protein is then tilted about the y-axis.
- The (initial) rotation axis should be along the long axis of the protein, by default this is assumed to be the z-axis. About this axis, the protein is rotated with the rotation interval until a foll rotation has been carried out. Then the protein and the rotation axis are tilted away about the tilt axis and tilt interval and the molecule is rotated about the new rotation axis. This is repeated until the maximum tilt angle has been reached.
The protein is automatically moved to the origin of the coordinate system before
the orientation search and a report is produced as a PDF file. This report visualizes
the initial orientation of the protein with the tilt and rotation axes and summarizes
the results. It includes plots of the protein at each tilt angle along with a comparison
plot of the spectra for each rotation at this tilt and 3D graphic showing the
change of intensity during the rotation. It is also supposed to help understanding the
naming convention of the files (jobname.t120.r030.cd for a tilt angle of 120 degrees and
a rotation about 30 degrees).
Job Submission
Mail Address
Some of the calculations, especially when charge-transfer transitions are considered, can take between half an hour and longer. Hence, the results are generally only provided by mail.
The output files are all plain text (including the plots which are postscript files) and usually not very large and in generall compressed as zip archive. Unless a big number of PDB files is processed, the returned archive is small enough to be sent by mail.
Download link
If the compressed folder with the results of the job is bigger than 10 MB, a download link is sent via mail instead of the archive itself. If even mails smaller than 10 MB are unwanted or the mail server blocks the DichroCalc mails, the sending of a download link can be forced with this tickbox.
Protein Sizes and Compiler Limits
The most expensive step of the calculations is the diagonalization of the Hamiltonian matrix. For each transition on every chromophore the dimension of this matrix increases by one. The maximum values, which can be handled by the program, are:
- 2,000 groups/chromophores
- 10,000 atoms
A protein with 501 residues contains 500 peptide bonds and hence the number of chromphores would be 500 for the plain calculation. Adding charge-transfer increases the number about 250 (since two peptide groups form a charge-transfer chromophore). Considering side chain chromophores adds a varying number of groups depending on the respective protein.
In some cases, the size limitations can be circumvented. If the protein comprises multiple chains then DichroCalc will automatically split the PDB into these and – provided that the chains do not exceed the limits on their own – will perform the calculation for each of them. Many proteins consist of multiple chains with identical sequence and, therefore, identical secondary structure. If the single chain spectra are similar (a comparison plot is generated), then the whole protein resulted in the same spectrum if it would be possible to calculate it. However, this is only possible for CD since in LD calculation the orientation of the chains is also important.
File Types
Choosing one of the four checkboxes is mandatory as otherwise no calculation is performed. For both a CD and LD calculation, line spectra are calculated first and are then superimposed with bands of the selected curve type and chosen wavelength to produce the curves.
.cdl | CD line spectrum |
.cd | CD spectrum |
.abl | Absorbance line spectrum |
.ab | Absorbance spectrum |
.ld | LD spectrum |
.dr | Dichroic Ratio spectrum |
.ldr | reduced LD spectrum |
.exp.cd | Experimental CD spectrum |
.exp.ld | Experimental LD spectrum |
.exp.dr | Experimental Dichroic Ratio spectrum |
.exp.ldr | Experimental reduced LD spectrum |
Results
The calculation of a CD spectrum for a protein with around a 1000 residues takes only a couple of minutes, if only the backbone peptide bonds are considered. Side chain transitions prolong the calculation time only slightly. However, if charge-transfer transitions are taken into account, the processing time can be up to half an hour per protein.
The user is sent an email with z zip archive attached which expands into a folder containing the results of the job. These are the xy files containing the spectra for the use in a spreadsheet application like Excel, Origin or Numbers (see File Types), postscript files with the plots of the spectra and the log files.
The Log Files – What to Check After the Calculation
There are two different log files. For the overall job the .log file contains a
summary of all files that have been calculated. The base name of the file is the
Job Name. It contains information when the job had been calculated and all selected
parameters:
Job sent: Tuesday, 21/10/2008 - 16:17 File name: 5wez.pdb Backbone chromophore: Ab initio parameter set (Hirst et al.) Selected curve type: Gaussian shape Range: 150 to 250 nm every 0.1 nm Selected bandwidth: 12.5 nm Units: Ellipticity Send CD spectrum: YES Send CD line spectrum: NO Backbone transitions: 2 Charge-transfer transitions: NO Include cyclic peptide bonds: NO Aromatic side chain transitions: NO Include asparagine side chain: NO Include glutamine side chain: NO Send absorbance line spectra: NO Send absorbance spectra: NO Send LD spectrum: NO Send dichroic ratio spectrum: NO Send reduced LD spectrum: NO Orientation search: NO
After some status messages (which are mainly important if something went wrong and one needs to find out at which point) it states how many files are processed and whether the individual calculations have been successful or failed:
Beginning processing of 3 files ==================================================================================================== Calculation successful: 5skw.pdb Calculation successful: 2ath.pdb Calculation successful: 6aww.pdb Processing finished Successful files: 3 Failed files: 0 ====================================================================================================
In the list above it would be directly obvious if a job had failed. In this case it can be checked
in the respective .log file what went wrong. For each file that is processed on DichroCalc
a .log is returned containing detailed information about the calculation.
The header of a .log file contains the same information as given in the job summary (
.job), all selected parameters which have been selected by the user.
The following section give information gathered during the course of the calculation:
- Creation of input files for the calculation
Errors here usually point to problems with the PDB format. The program detects the chains in the file (also chain breaks which may be due to missing residues or abnormal residue distances) and assigns the selected chromophores such as peptide bonds and aromatic side chain groups. For this routine it is important that the PDB atom labels are correct.
assigned chromophores --------------------- 85 peptide chromophores 84 CT chromophores 3 PHE chromophores 4 TYR chromophores 2 TRP chromophores
The assigned atom assignments are then given for each found chromophore:Assignments of the atoms ------------------------ The numbers refer to the renumbered PDB, not the original! Atom assignments for peptide groups: 3 4 7 9 10 16 18 19 21 ...Each line represents one assigned chromophore starting with the peptide groups. The first peptide bond is assigned to the atoms 3, 4 and 7, which must be the PDB atom labels C, O and N (see PDB atom labels). Since the PDB file has to be renumbered in the process, these number do not necessarily match with the original atom numbers. The PDB information after renumbering is given in between the markers
==PDBSTART==and==PDBEND==:Renumbered PDB input file: ==PDBSTART== ATOM 1 N SER A 1 13.243 1.352 9.718 1.00 54.86 N ATOM 2 CA SER A 1 14.014 0.398 8.869 1.00 54.36 C ATOM 3 C SER A 1 15.389 0.125 9.469 1.00 53.43 C ATOM 4 O SER A 1 16.079 1.048 9.906 1.00 53.69 O ATOM 5 CB SER A 1 14.184 0.964 7.454 1.00 54.66 C ATOM 6 OG SER A 1 12.929 1.201 6.840 1.00 54.95 O ATOM 7 N GLN A 2 15.777 -1.147 9.489 1.00 52.51 N ATOM 8 CA GLN A 2 17.076 -1.549 10.019 1.00 50.78 C ATOM 9 C GLN A 2 18.153 -1.248 8.982 1.00 49.03 C ATOM 10 O GLN A 2 19.304 -0.971 9.326 1.00 47.68 O ATOM 11 CB GLN A 2 17.072 -3.046 10.346 1.00 51.07 C ATOM 12 CG GLN A 2 18.438 -3.637 10.693 1.00 53.44 C ATOM 13 CD GLN A 2 19.066 -3.013 11.926 1.00 55.72 C ATOM 14 OE1 GLN A 2 18.427 -2.899 12.974 1.00 57.60 O ATOM 15 NE2 GLN A 2 20.328 -2.617 11.811 1.00 56.27 N ... ===PDBEND==
- Dichroism calculation
This section may contain errors if the file was too large (too many chromophores or atoms) to be processed. If the single chains are small enough then each chain is calculated individually for CD and a comparison plot of the single chain spectra is generated. Often the chains are almost identical and the spectra will match. - Addition of bandshapes to the line spectra
This section may contain warnings about negative wavelengths. These are usually due to problems in the structure itself, for example, chromophores being too close like in abnormally tight turns. The structure should then be checked for such problems. - Plot of CD/LD spectrum
Spectrum Plots and XY-Data
The calculated spectra are plotted on their own and - if provided - in comparison with the experimental spectrum. Both the postscript files and the xy-data textfiles are compressed in a zip archive which is returned to the user. When the calculation is finished an email is sent to the given address, informing about how many files were processed and how many of them had failed. If the archive is smaller than 10 MB, it is attached to the mail, in case of it being larger than 10 MB, a link is sent where it can be downloaded. This behaviour can also be forced by selecting the respective option in the form.